GMP认证申请条件
GMP验厂咨询:不是任何企业都可以申请GMP认证,企业在申请定要做一些验厂咨询,多了解一些有关GMP验厂的知识,或者找的验厂辅导公司,做一次验厂辅导。
企业要申请GMP认证,具备下列5个条件:
(一)领有公司执照或商号之营利事业登记证者。
(二)领有经济部工厂登记证,并载有申请认证之产品项目者。
(三)符合现行食品有关法令、食品工厂GMP通、专则及本规章有关规定者。
(四)从事分装或改装产品之厂商,得申请食品GMP认证,惟其分装或改装前之产品应先取得食品GMP认证。
(五)加工层次轻微之产品,得视同分装或改装产品。如判定困难时,得提请食品GMP技术会研议。
以上5点是企业具备的,也是GMP验厂通过的先决条件。
创思维的服务宗旨“辅导+良好人脉”双确保工厂通过验厂,专为工厂解决验厂中遇到的各种难解之题;强大的顾问团队,的服务口碑,验厂关系通全国、可提前安排审核,现场、审核时间查询、率; 选择创思维-任何原因导致验厂不通过,我们负责到底!
GMP认证新版
根据共和国部长签署的2011年第79号令,《品生产质量管理规范(2010年修订)》(下称新版GMP)已于2010年10月19日经部务会议审议通过,自2011年3月1日起施行。
新版GMP与98版相比从管理和技术要求上有相当大的进步。特别是对无菌制剂和原料的生产方面提出了很高的要求,新版GMP以欧盟GMP为基础,考虑到国内差距,以WHO2003版为底线。
新版GMP认证有两个时间节点:品生产企业血液制品、、剂等无菌品的生产,应在2013年12月31日前达到新版品GMP要求;其他类别品的生产均应在2015年12月31日前达到新版品GMP要求。未达到新版品GMP要求的企业(车间),在上述规定期限后不得继续生产品。
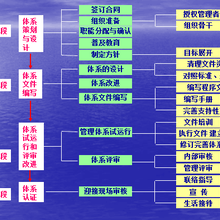
GMP认证审核程序
1 、职责与权限
1.1省、自治区、直辖市品监督负责该辖区品生产企业品 GMP 认证申报资料的初审及日常监督管理工作。
2 、认证申请和资料审查
2.1 申请单位须向所在省、自治区、直辖市品监督管理部门报送《品 GMP 认证申请书》,并按《品 GMP 认证管理办法》的规定同时报送有关资料。省、自治区、直辖市品监督管理部门应在收到申请资料之日起 20 个工作日内,对申请材料进行初审,并将初审意见及申请材料报送品监督司。
2.2 认证申请资料经局司受理、形式审查后,转交局认证中心。
2.3 局认证中心接到申请资料后,对申请资料进行技术审查。
2.4 局认证中心应在申请资料接到之日起 20 个工作日内提出审查意见,并书面通知申请单位。
3 、制定现场检查方案
3.1 对通过资料审查的单位,应制定现场检查方案,并在资料审查通过之日起 20 个工作日内组织现场检查。检查方案的内容应包括日程安排、检查项目、检查组成员及分工等。在资料审查中发现并需要核实的问题应列入检查范围。
3.2 局认证中心负责将现场检查通知书发至被检查单位,并抄送其所在地省级品监督管理部门、检查组成员所在单位和局司。
3.3 检查组一般不超过 3 人,检查组成员须是品监督品 GMP 检查员。在检查组组成时,检查员应回避该辖区品 GMP 认证的检查工作。
4 、现场检查
4.1 现场检查实行组长负责制。
4.2 省级品监督管理部门可选派一名负责品生产监督管理的人员作为观察员参加辖区品 GMP 认证现场检查。
4.3 局认证中心负责组织 GMP 认证现场检查,并根据被检查单位情况派员参加、监督、协调检查方案的实施,协助组长草拟检查报告。
4.4 会议内容包括:介绍检查组成员;声明检查注意事项;确认检查范围;落实检查日程;确定检查陪同人员等。检查陪同人员是企业负责人或生产、质量管理部门负责人,熟悉品生产全过程,并能准确解答检查组提出的有关问题。
4.5 检查组须严格按照检查方案对检查项目进行调查取证。
4.6 综合评定检查组须按照检查评定标准对检查发现的缺陷项目进行评定,作出综合评定结果,拟定现场检查的报告。评定汇总期间,被检查单位应回避。
4.7 检查报告须检查组全体人员签字,并附缺陷项目、尚需完善的方面、检查员记录、有异议问题的意见及相关资料等。
4.8 未次会议检查组宣读综合评定结果。被检查单位可安排有关人员参加。
4.9 被检查单位可就检查发现的缺陷项目及评定结果提出不同意见及作适当的解释、说明。如有争议的问题,必要时须核实。
4.10 检查中发现的不合格项目及提出的尚需完善的方面,须经检查组全体成员及被检单位负责人签字后,双方各执一份。
4.11 如有不能达成共识的问题,检查组须作好记录,经检查组全体成员及被检单位负责人签字后,双方各执一份。
5 、检查报告的审核
局认证中心须在接到检查组提交的现场检查报告及相关资料之日起 20 个工作日内,提出审核意见,送品监督司。
GMP认证后的营销走向
1 、重视开拓海外市场
制工业 GMP 认证的完成,给制企业海外市场带来一片曙光,尤其是中制企业,无论是大中小型制企业都将面临同一平台上的竞争,营销工作相对容易许多,尤其是东南亚市场的状况。东盟有使用中的传统,如果其他承认的 GMP 标准,过了 GMP 就相当于有了准入证,而且海外市场竞争不激烈。
2 、通过 OEM 生产,消化闲置产能
对于国内企来讲,进行 OEM 代加工生产,是消化闲置产能一个重要途径。品委托生产在国外很普遍,可以合理使用社会资源,我国修订后的《品管理法》也对此作了明确规定。
企业可以主动寻求和国内一些大制企业合作,成为他们的生产代工车间,赚取生产利润。中小企业因无新产品跟进, GMP 后面临开工不足的困境。可以南北合作,北方的企业帮助南方的企业委托加工,南方的企业协助北方的企业 OEM ,或者东西合作,这样大家的运输成本都会大幅度降低,尤其是对于销售半径不大的较重的水剂、颗粒剂很适合用此法。 企业还可以纵向联合 OEM :解决流通企业的 OEM 冲动。医流通企业对品销售市场的动态及未来趋势都能够比较好的把握,知道什么产品能够为企业带来利润,但往往却为寻找这样的产品而花费的精力,而且生产商也以 " 全国 "" 国准字 "" ×类新 " 等为法码,提高产品出厂价格,压榨代理商的利润,让代理商处境维艰。因此他们迫切想开发具有自己知识产权的产品。但他们由于没有 GMP 厂房,无法立项,这给制企业带来契机。完全可以与这些企业合作研发产品,为其生产产品。
国内企还要注意和国外制跨国公司的联合,为其 OEM 。廉价的劳动力和相对廉价的生产成本,完全可以为外资企在做品的外包生产,但原来厂较落后的生产工艺和不稳定的产品质量很大程度上妨碍了这一模式。 GMP 认证为外资企在的生产外包扫除了障碍。江浙等地的制企业已经开始这一尝试,如浙江海正为美国默克、苏州立达为美国惠氏的品加工等等。制企业已悉数进入,相信这应是一个较大的市场。
药品GMP认证是国家依法对药品生产企业(车间)和药品品种实施GMP监督检查并取得认可的一种制度。虽然国际上药品的概念包括,但只有中国和澳大利亚等少数几个国家是将人用药GMP和GMP分开的。
办理程序:
(一)申请:
申请人向省政务大厅食品药品监督管理局窗口提交《药品GMP认证申请书》(一式两份)及申请书电子文档,并附相关材料。
(二)受理:
申请材料经省政务大厅食品药品监督管理局窗口形式审查符合要求的,予以受理,出具受理通知书;申请材料不或者不符合形式审查要求的,5个工作日内发给申请人《补充材料通知书》;不予受理的出具《不予受理通知书》。
(三)现场检查:
经形式审查符合要求的转入技术审查,技术审查符合要求的安排现场检查。技术审查需要补充材料的,一次性书面通知申请企业。企业应在2个月内报送,逾期未报的中止认证工作。
(四)审查:
省食品药品监督管理局对现场检查报告进行审核和审批,符合认证标准的报国家食品药品监督管理局予以公告,公告无异议的,颁发《药品GMP认证审批件》和《药品GMP证书》,公告有异议的,组织调查核实。对不符合药品GMP认证标准要求的,且无法通过限期改正达到标准的,发给《药品GMP认证审批意见》;可以责令企业限期改正的,向被检查企业发整改通知书,整改的时期为6个月。企业在期限内改正完毕,提交改正报告,符合要求的派检查组再次进行现场检查。经再次现场检查,符合药品GMP认证标准,颁发《药品GMP认证审批件》和《药品GMP证书》;仍不符合的,发给《药品GMP认证审批意见》。
药品生产企业被吊销、缴销《药品生产许可证》或者被撤消、注销生产范围的,其相应的《药品GMP证书》由原发证机关收回。